
This module of MestReNova is an efficient simulator for high resolution NMR spectra.
Follow the menu ‘View/Panels/Spin Simulation’ or ‘Advanced/Spin Simulation’ which will display the corresponding dialog box:

Simulate a spectrum by entering the desired values. For example, imagine 3 protons (3 spins Groups) called A, B and C, with chemical shifts 1, 5 and 7 ppm respectively. Their coupling constants are J(AB) = 3Hz, J(BC) = 5.5Hz and J(CA) = 3.4Hz. Finally select the number of points and the spectral limits and click on the ‘New’ button to generate the corresponding spectrum:
Once the spectrum is simulated, modify any value and obtain the new spectrum by clicking on the ‘Recalculate’ button. You can introduce ‘Dipolar’ or ‘Quadrupolar’ coupling constants and change the ‘number of nuclei’, ‘Spin’ and the ‘Line Width’ values by clicking and entering the desired values on the corresponding boxes. Finally click on the ‘Recalculate’ button to update the changes in the spectrum
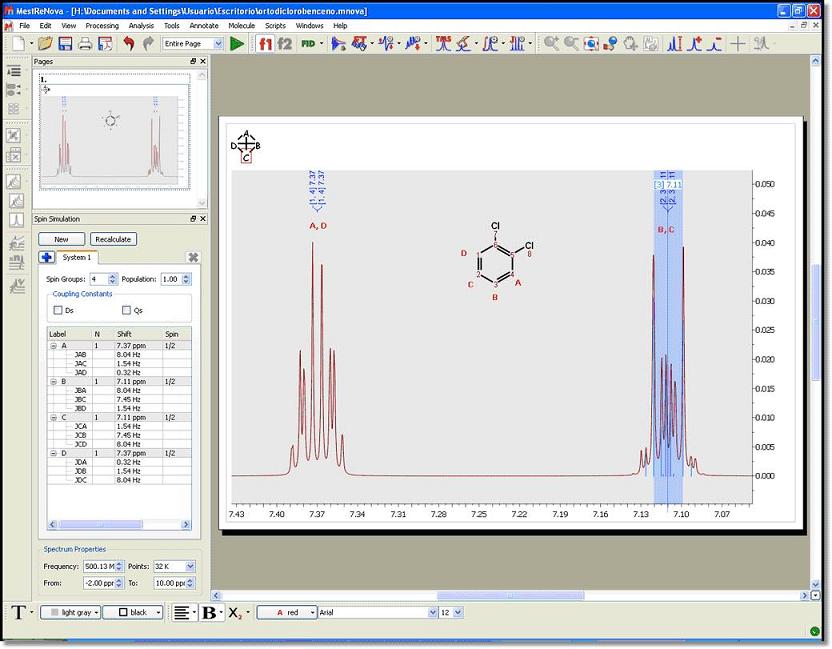
You can see below the simulated spectrum of the ‘orto-dichlorobencene’; which is a AA’BB’ system:
You can add several systems just by clicking on the ‘Add System’ button . A new system will be a fragment of the molecule that can be simulated separately since its protons do not interact with the rest. In other words, a set of nuclei that are related through their coupling constants. Once you have added a new system, you can change the population of the system by using the ‘Population’ edit box.
As an example, consider the case of a small molecule, ethyl acrylate CH2CHCOOC2H5
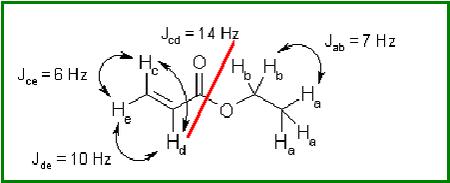
As you can see, a standard simulation of the 1H-NMR spectrum of this molecule would require eight frequencies to be introduced, one for each proton. Moreover, you should introduce nine coupling constants as well. However, even a brief study of this spin system suggests us a more elegant solution
Why not treat this spin system as two ‘subsystems’?
One ‘subsystem’, corresponding to the ethyl group, is composed of five protons with only two distinctive frequencies labeled a (the methyl group) and b (the methylene group), and only one coupling constant, Jab. Other ‘subsystem’ is composed of the three remaining protons (those of the vinyl group), with three frequencies, c, d and e, and three coupling constants, Jcd, Jce and Jde. This decomposition of the spin system in two subsystems is possible because they are not connected in any way. The decomposition of a spin system into subsystems can be carried out in many molecules, and is, for example, the conceptual basis of the NMR spectroscopic data tables.
See below, the synthesized spectrum of the ethyl acrylate; after having created two different systems; system A (vinyl protons) in red, and system B (ethyl group) in green:
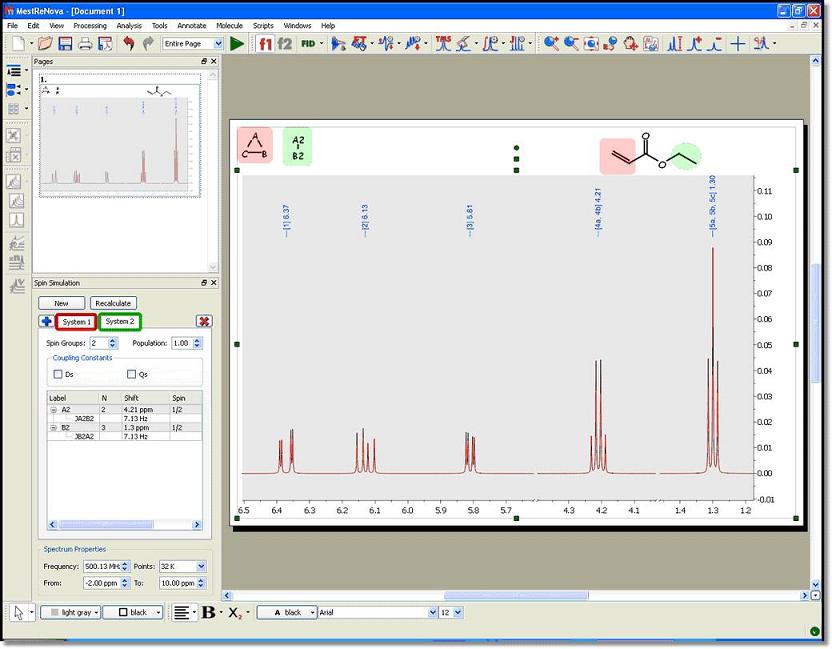
This decomposition method represents a great advance in terms of calculation speed, because the complexity of the calculation grows exponentially with the number of spins (nuclei) considered. However, we can perform even faster calculations and achieve greater flexibility going one step further.
Why not introduce only five frequencies, and tell the program how many protons correspond to each frequency?
The introduction of magnetic equivalence allows the calculation to be performed using five spin groups, labeled from a to e, with their corresponding frequencies. The two first spin groups, a and b, have three and two protons respectively; while the other three spin groups have only one proton each. Of course, we need then only four coupling constants, Jab, Jcd, Jce and Jde. This process can be carried out in many molecules, and is more intuitive than the classical one.
")

